

Cuadernos de Información Económica, N.º 303
(noviembre-diciembre 2024)
Fecha: noviembre 2024
Autor
Félix Lobo*
Etiquetas
Sector farmacéutico, reforma legislativa europea, I+D+i, medicamentos innovadores
Los incentivos a la I+D+i en la reforma de la legislación europea del medicamento
Este artículo aborda la reforma legislativa europea en el sector farmacéutico, destacando el papel de la I+D+i en medicamentos innovadores, especialmente en enfermedades raras y nuevas terapias. La Comisión Europea propone ajustes en la exclusividad de mercado y en la protección de datos para equilibrar la innovación y la accesibilidad. Estos incentivos buscan incentivar lanzamientos más rápidos y asequibles en todos los Estados miembros, mientras que los elevados costos de algunas terapias plantean preocupaciones sobre la sostenibilidad del sistema sanitario. Se examina también el impacto de la legislación en la competitividad europea frente a EE. UU. y Asia.
Introducción
Como dice la Estrategia Farmacéutica para Europa, “la UE cuenta con una industria farmacéutica sólida y competitiva. Junto con otros actores públicos y privados, sirve a la salud pública y actúa como motor de la creación de empleo, del comercio y la ciencia. Los fabricantes de medicamentos hicieron en 2019 la mayor contribución a la inversión en investigación, con más de 37.000 millones de euros. El sector ofrece 800.000 empleos directos y un excedente comercial de 109.400 millones de euros” (Comisión Europea, 2020a). Desde la Economía del Desarrollo y la Historia Económica, el Nobel Deaton juzga que los medicamentos “han salvado millones de vidas… y permitido a millones de personas… continuar trabajando, percibiendo ingresos y amando…” (Deaton, 2015: 159).
En la actualidad asistimos a una oleada innovadora. En 2023 se autorizaron setenta nuevos medicamentos por las agencias reguladoras europea, estadounidense o inglesa (Papapetropoulos et al., 2024). En el gráfico 1 se puede apreciar esta dinámica en los últimos veinte años.
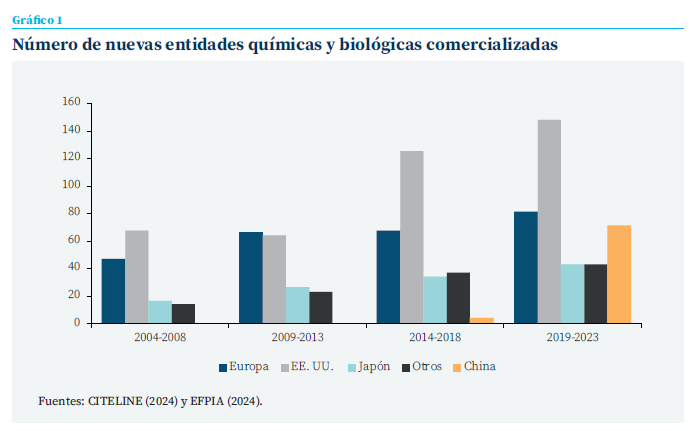
Esta oleada está protagonizada, sobre todo, por los nuevos productos biológicos (proteínas recombinantes o anticuerpos monoclonales), la inmunología y los oncológicos. Entre ellos, muchos son destinados a enfermedades raras, cánceres sin tratamiento, o productos para terapias avanzadas (Papapetropoulos et al., 2024). Baste señalar como ejemplos de innovaciones recientes, los tratamientos de la hipertensión y enfermedad cardiovascular, esclerosis múltiple y migraña, antirretrovirales para VIH-SIDA, los antivirales de acción directa que curan la hepatitis C, los agonistas GLP-1 para diabetes y obesidad, vacunas contra la COVID-19, virus respiratorio sincitial y herpes zóster; anticuerpos monoclonales para varias enfermedades autoinmunes y oncológicas y las muy prometedoras terapias avanzadas (génicas y celulares), entre ellas la terapia de células T con receptores quiméricos de antígenos (CAR-T), o las terapias CRISPR-Cas9 basadas en tecnología de edición del genoma, así como los medicamentos huérfanos (MH) para muchas enfermedades raras.
Las políticas estatales de fomento de la innovación farmacéutica y los incentivos que articulan han sido y siguen siendo de la mayor importancia, ya que en condiciones de mercado no regulado sabemos que la I+D+i sería insuficiente para cubrir las necesidades sociales
En estos desarrollos, las políticas estatales de fomento de la innovación farmacéutica y los incentivos que articulan han sido y siguen siendo de la mayor importancia, ya que en condiciones de mercado no regulado sabemos que la I+D+i sería insuficiente para cubrir las necesidades sociales. Las patentes son el ejemplo más notorio, pero aquí no nos vamos a referir a ellas, sino a tres políticas europeas complementarias o adicionales peculiares del sector farmacéutico, que son calificables como “políticas tirón” (pull), en cuanto aumentan los beneficios esperados (Kyle, 2022), y que complementan o conceden formas de monopolio o exclusividad de mercado1. La “Protección de Datos Regulatoria” significa que los estudios científicos presentados por el innovador para obtener la autorización de comercialización y sus datos no pueden ser utilizados para fundamentar la solicitud de un genérico o un biosimilar potencial competidor del original, lo cual eleva una barrera de entrada muy eficaz porque estos estudios son costosos. (En la UE dura ocho años actualmente, más uno si se obtiene autorización para una indicación nueva significativa de una sustancia ya conocida). La así llamada en la legislación europea “Protección del Mercado o comercial” (dos años adicionales a la protección de datos), significa que el mercado –el grupo de pacientes al que va destinado el medicamento– se reserva exclusiva- mente al titular del medicamento beneficiario, por el hecho de ser autorizado. En ambos casos, la protección empieza a contar desde la autori- zación de comercialización.
Otra política de “tirón” de la oferta es la exclusividad especial concedida a los “medicamentos huérfanos”, esto es, los destinados a enfermedades raras, es decir, las que afectan a colectivos pequeños, en la UE a menos de cinco de cada 10.000 habitantes. Sin medidas estatales de fomento, en principio tienen poco interés comercial en condiciones de mercado, por su escasa rentabilidad. En EE. UU. y en la UE la legislación (Orphan Drug Act de 1988 y Reglamento (CE) n.º 141/2000), tras una calificación formal como tales medicamentos huérfanos y la autorización de comercialización, los beneficia con incentivos sustanciales, consistentes básicamente en una potente protección de mercado que dura siete y diez años respectivamente2 (no adicionales sino por todos los conceptos), además de otros beneficios regulatorios tipo “empujón a la oferta”. Algunos medicamentos huérfanos han demostrado gran eficacia, ya que consiguen curar o paliar enfermedades raras muy graves.
Obsérvese que la protección de datos y la protección del mercado son independientes de las patentes, de modo que pueden coexistir o pervivir cuando aquellas han expirado, y a la inversa. Además, son derechos de exclusiva más fuertes que las patentes. Estas tienen cierta incertidumbre en cuanto a su duración3 y se ven condicionadas por una serie de requisitos de cierta complejidad, por lo que son campo abonado para la litigiosidad que puede resultar en su anulación. En cambio, estas exclusividades especiales del mercado farmacéutico no son inciertas sino automáticas, pues únicamente dependen de obtener la autorización (sanitaria) de comercialización. Otra diferencia importante es que el monopolio que conceden es radical, más fuerte que las patentes, pues reservan el propio mercado, la subpoblación de pacientes y no sólo protegen la sustancia. Es decir, mientras duran, una empresa potencialmente competidora no puede entrar en dicho mercado ni con un genérico, ni con un biosimilar, ni tampoco con un medicamento basado en una sustancia alternativa similar (un “me-too”), cosa que sí permite la protección de patentes. Solamente podría hacerlo si el producto alternativo supone una mejora. Se trata también de condiciones de monopolio a priori duraderas.
Sin embargo, estos nuevos tratamientos tienen frecuentemente elevados costes, con cifras de cinco y seis dígitos en euros por paciente. El tisagenlecleucel (Kymriah®), la primera de las novedosas terapias CAR-T, fue incluido en la prestación farmacéutica del Sistema Nacional de Salud español en 2018 a un precio de 320.000 euros (aunque este es un precio “de catálogo” y está sujeto a acuerdos especiales de riesgo compartido) (Lobo, 2019). En 2023 en EE. UU. el récord de precio lo alcanzó una terapia génica para la hemofilia B, Hemgenix (etranacogene dezapar- vovec), lanzada con un precio de catálogo de 3,5 millones de dólares USA (Horrow y Kesselheim, 2023). Estos elevados costes causan preocupación por la sostenibilidad de los sistemas sanitarios.
Especialmente en el caso de los medicamentos huérfanos, se discute, como luego veremos, si el orden de prioridades implícito en estas políticas frente a otras opciones, es el correcto.
Estos elevados costes causan preocupación por la sostenibilidad de los sistemas sanitarios. Especialmente en el caso de los medicamentos huérfanos
En este marco, la Comisión Europea detectó como problemas actuales y prioritarios del mercado farmacéutico de la Unión los siguientes (Comisión Europea, 2023a):
- Los medicamentos autorizados en la UE no llegan a los pacientes con la suficiente rapidez y no son accesibles por igual en todos los Estados miembros (EE. MM.).
- Existen lagunas importantes al abordar las necesidades médicas no satisfechas, las enfermedades raras y los nuevos antimicrobianos.
- Los precios tan altos.
- Los problemas de suministro de medicamentos (escaseces).
- Las cargas administrativas.
- El impacto medioambiental de la fabricación.
Para solucionar estas dificultades se está tramitando una importante reforma de la regulación europea que pasamos a describir.
Descripción de la reforma legislativa en marcha
Los objetivos de esta reforma legislativa son los siguientes:
- Disponibilidad de los medicamentos por todos los pacientes de la UE.
- Favorecer la I+D+i, manteniendo un equilibrio con la accesibilidad por los sistemas de salud y los pacientes en condiciones asequibles.
- Mejorar la asequibilidad de los medicamentos mejorando la cooperación entre los EE. MM. en precios y financiación pública y adquisiciones comunes, así como facilitando la entrada en el mercado de medicamentos genéricos y biosimilares.
- Seguridad de los suministros.
- Fomento de la producción de medicamentos en Europa.
- Protección del medioambiente.
- Abordar las resistencias a los antimicrobianos.
- Acelerar y simplificar los trámites y apoyar a las empresas solicitantes de autorización de comercialización tempranamente (Comisión Europea, 2020a y 2023a).
En este artículo solamente nos centramos en las propuestas de modulación de los incentivos a la innovación y su equilibrio con la accesibilidad. La propuesta de nueva legislación europea pretende sustituir la regulación incondicionada de la protección de datos y la exclusividad de mercado por una regulación condicionada a ciertos objetivos con periodos e incentivos distintos según los casos. La propuesta no afecta a las patentes (Comisión Europea, 2023a;, 2023b y 2023c).
Como se ve en el gráfico 2, el sistema actual la protección abarca en general un máximo de 11 años (8 + 1 + 2) y 12 (10+2) años para los medicamentos huérfanos.
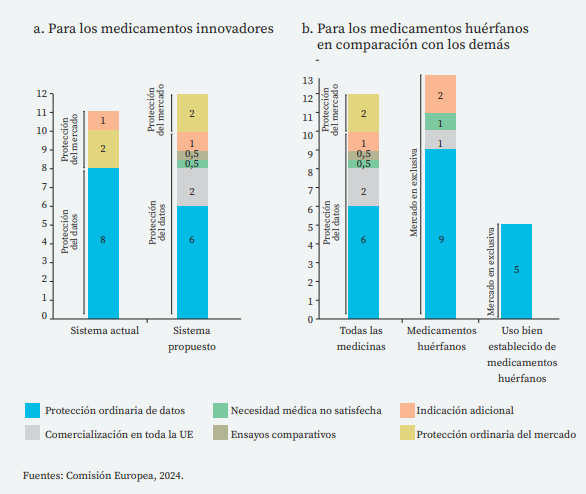
Según la propuesta de la Comisión el periodo de protección podría alargarse hasta 12/13 años, pero en función de los objetivos:
- Habría un mínimo de seis años de protección de datos y otros dos años de protección de mercado para los medicamentos innovadores (9 para MH por ambos conceptos).
- Dos años adicionales de protección de datos (1 para MH) si se comercializa el pro- ducto en todos los países de la UE antes de trascurridos dos años desde la autorización europea (tres si es una pyme o una entidad sin fines de lucro).
- Medio año más si el medicamento resuelve una necesidad médica no cubierta (un año si es un MH y ha de ser una necesidad elevada).
- Medio año adicional de protección de datos si se presentan ensayos clínicos comparativos (no para MH).
- Un año más de protección de datos para las indicaciones adicionales (para los MH hasta un máximo de dos años).
En el caso de los MH la regla general de exclusividad de mercado incondicional pasa de los 10 años actuales a 9 años, pero solamente a cinco si la solicitud de autorización de comercialización se basa en literatura científica (medicamento suficientemente conocido) más las extensiones condicionales dichas.
Algunas reflexiones desde la economía
En primer lugar, nos podemos preguntar si la legislación europea con objetivos sanitarios (garantizar la eficacia, seguridad y calidad de los medicamentos) es el marco adecuado para los incentivos a la innovación y si no sería más conveniente regularlos y administrarlos en el marco normativo de la propiedad intelectual, para una mayor coherencia. A este respecto se ha recordado el principio de Tinbergen: cuando se persiguen diferentes objetivos de política económica conviene asignar una medida para uno de ellos, para que se puedan controlar y no se contradigan (Castanheira y Siotis, 2023). Pero también es cierto que la propuesta de reforma diversifica los incentivos según distintos objetivos, con lo cual nos estaríamos acercando a la regla de Tinbergen (Ibíd.).
Una preocupación europea es la medida en la que la innovación tiene lugar en Europa o se está desplazando hacia EE. UU., Japón o China. Mientras que en 2004-2008, Europa era capaz de generar el 33 % de los medicamentos innovadores, en 2019- 2023 generaba el 21 % (EFPIA, 2024) (aunque estos datos no están filtrados según grado de valor terapéutico de los productos). Los incentivos que excluyen temporalmente la competencia en precios pueden tener efectos positivos sobre la innovación globalmente. Pero eso no significa que la legislación de una determinada región garantice ni estimule la innovación en esa región concreta. En la Unión Europea la regulación se aplica igualmente a todas las empresas que venden en el mercado europeo, con independencia del origen de la compañía o el lugar en el que se desarrollan las actividades de I+D+i. Como puso de relieve Becker (2024), una exclusividad de un año más en una región determinada puede que genere ingresos más altos por ventas en dicha región. Esto puede estimular la innovación global, pero no necesariamente en la región concreta que promulga dicha legislación. A la inversa, un año menos de protección no significa que la investigación sea deslocalizada de esa región. La localización de las I+D+i está determinada por factores diversos, tales como políticas fiscales, disponibilidad de científicos y técnicos, facilidades de acceso al capital, inversiones públicas, universidades punteras. La realidad europea proporciona un ejemplo. En 2004 la legislación farmacéutica europea aumentó los periodos de exclusividad a diez años (8+2), mientras que con anterioridad oscilaba entre seis y diez años, dependiendo del Estado miembro. Sin embargo, el gasto empresarial en I+D+i no aumentó (Becker, 2024).
En 2004 la legislación farmacéutica europea aumentó los periodos de exclusividad a diez años (8+2), mientras que con anterioridad oscilaba entre seis y diez años, dependiendo del Estado miembro. Sin embargo, el gasto empresarial en I+D+i no aumentó
Un gran tema actual de discusión es en qué medida los incentivos en favor de los medicamentos huérfanos (MH) son eficientes y si implican un orden de prioridades correcto frente a otras opciones, según efectividad, coste y poblaciones afectadas. Podrían estar provocando un efecto desplazamiento de otras terapias o intervenciones, que podrían ser más eficientes (coste-efectivas) y compiten por los recursos disponibles con otros factores, como la incorporación de médicos y enfermeras. Por definición, un medicamento huérfano en los EE. UU. beneficia a menos de 200.000 personas y en la UE al 5 % o menos de la población. La Agencia Europea del Medicamento (EMA, por sus siglas en inglés) ha estimado que las más de 6.000 enfermedades raras afectan a uno de cada doce habitantes de la UE (6 %, 36 millones de personas) (EMA, 2023).
En general, los estudios empíricos muestran que, en los países ricos, las actividades de I+D responden positivamente a las expectativas de beneficios incrementados y a los refuerzos de la protección de la exclusividad (Kyle, 2022). Gaessler y Wagner (2022) han hallado empírica- mente que la reducción del periodo de exclusividad impulsa significativamente al abandono de los proyectos de investigación. En el caso de los MH parece que los beneficios económicos que han generado han sido sustanciales. Castanheira y Siotis (2023) resumen investigaciones empíricas que muestran que el valor presente del flujo de ingresos esperados de un MH es comparable al resto de los medicamentos y los han calificado de nueva “fiebre del oro”. Estos beneficios han atraído la inversión y se han desarrollado, autorizado y comercializado en gran número. Como se ve en el gráfico 3, suponen alrededor de un tercio de todas las autorizaciones de nuevos medicamentos. Desde la entrada en vigor de la regulación europea en 2000, la EMA ha calificado como huérfanos 2.870 sustancias en proceso de investigación y autorizado 240 MH (EMA, 2023), algunos de gran eficacia. En EE. UU. la evolución es similar (Sarpatwari et al., 2018).
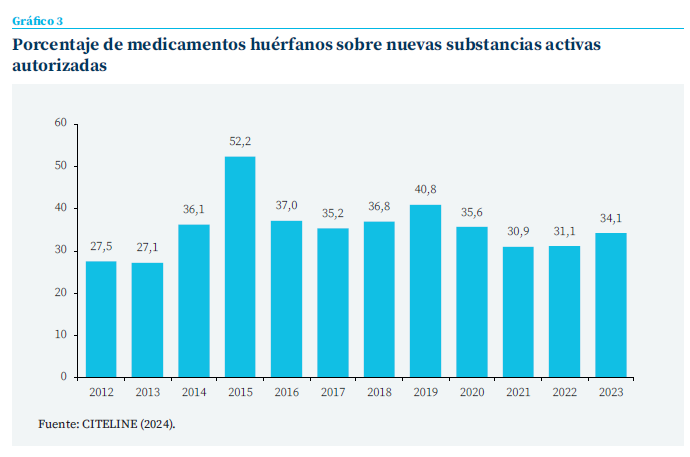
¿Podemos afirmar que estos incentivos legales, en forma de concesiones de monopolio, han tenido, por tanto, éxito? Comúnmente se estima que sí, aunque hay juicios menos favorables. Se pone de manifiesto que de las más de 6.000 enfermedades raras conocidas el 95 % no tiene actualmente ninguna opción de tratamiento (Comisión Europea, 2023a) después de 38 y 23 años de incentivos en EE. UU. y la UE. respectivamente, de modo que no habrían resuelto el problema para el que fueron establecidos (Castanheira y Siotis, 2023). El estudio de evaluación de la Comisión estimó entre 18 y 24, de un total de 142, los MH resultado directo de esta legislación y que ese total habrían beneficiado a unos 6,3 millones de pacientes (Comisión Europea, 2020b). Sarpatwari et al. (2018) restan importancia a estos incentivos, pues hallan que en EE. UU. en 1985-2014 solamente en el 33 % de los casos la exclusividad especial dura más que las patentes, por lo que en el resto su valor es discutible (aunque hay que recordar el efecto reductor de incertidumbre que tienen). Por otro lado, arguyen que por las características especiales de los MH no hay muchos estímulos para que los genéricos entren a competir con ellos, una vez que han caducado todas las protecciones.
Es claro que el desarrollo de los MH se debe no sólo a los incentivos legales, sino también al progreso científico y tecnológico, básicamente a los decisivos progresos en el conocimiento del genoma humano y las técnicas de ingeniería genética. La disponibilidad a pagar (altos precios) de los compradores institucionales (sistemas públicos de salud, aseguradoras) es un incentivo concurrente fundamental (Kyle, 2022). También se alega que la exclusividad podría haber sido redundante, pues se habrían desarrollado MH, incluso en ausencia de protección, dada la aceleración de la investigación en tiempos recientes en el sector (Castanheira y Siotis, 2023).
La Comisión Europea (2020b) ha estimado que en el 14 % de los casos, la exclusividad ha podido suponer una compensación excesiva y entonces no estaría justificada. Se trata de productos bien conocidos o medicamentos autorizados para múltiples indicaciones huérfanas. Este estudio de evaluación de la Comisión, por estas razones, considera que conviene un tratamiento diferencial de los incentivos y no “una talla única” para todos los casos, que es la visión gradualista que ha recogido la propuesta de reforma legislativa. Castanheira y Siotis (2023) han propuesto nuevos instrumentos de política novedosos.
Los costes que suponen para los sistemas de salud de los EE. MM. financiar los MH son otro dato relevante. Entre 2000 y 2017 totalizaron entre 20 y 25.000 millones de euros, aparte de los fondos públicos invertidos en investigación. Por otro lado, gracias a los MH los pacientes ganaron entre 210.000 y 440.000 años de vida ajustados por calidad (Comisión Europea, 2020b). En España, en 2021 el gasto en MH se estimó en 1.003,6 millones de euros, un 66,1 % más que en 2016, casi en su totalidad ejecutado por los hospitales, y supuso alrededor del 6 % del gasto público total en medicamentos. (DGCSSNSF, 2022).
La nueva regulación en trámite exige cumplir la condición de comercializar en todos los EE. MM. para ampliar de ocho a diez años la exclusividad, que es la situación actual. Según (Kyle et al., 2023) esta propuesta no tiene en cuenta algunas características importantes del mercado europeo relacionadas con la regulación. Los precios de lanzamiento de los nuevos productos farmacéuticos en la UE no son independientes entre países. Las empresas se ven afectadas en sus decisiones y calendario de lanzamiento por la presencia de los llamados “Precios de Referencia Externos” y por el comercio paralelo. Los “Precios de Referencia Externos” significan que los EE. MM. que intervienen los precios, (que son muchos), toman como referencia los precios aprobados por los demás países. Por ello, las empresas tienen incentivos a retrasar o incluso renunciar a la comercialización en los países con precios más bajos (en general, los de menor renta) (Maini y Pammolli, 2023). A ello se añade que un operador no titular de la patente de un producto puede legalmente exportarlo de un país de precios bajos a otro con precios altos y beneficiarse de la diferencia (arbitraje) aunque esté protegido y sin el consentimiento del titular. El comercio paralelo provoca una presión a la baja de los precios e incentiva a diferenciar productos entre países. En ausencia de estas dos restricciones (que derivan de regulaciones) las empresas tendrían incentivos a comercializar en cuantos más países mejor. El resultado es que los pacientes europeos pueden no acceder o beneficiarse más tarde de las innovaciones terapéuticas y que los productos disponibles varían entre países. Así, con la regulación actual, sólo el 12,8 % de los productos se lanzan simultáneamente en por lo menos veinte EE. MM. (Comisión Europea, 2023d).
La condición de comercializar en todos los EE. MM., según (Kyle et al., 2023) reduciría la rentabilidad de la innovación, la inversión en I+D y la competencia entre productos similares Los ingresos de las empresas que, a pesar de la nueva regulación, no comercializaran en todos los EE. MM., serían un 22 % inferiores a la situación actual (Comisión Europea, 2023d), pues solamente tendrían ocho años de exclusividad, en lugar de diez. Para las empresas que cambiaran su estrategia y sí comercializaran en todos los EE. MM., también habría pérdida de ingresos a pesar de mantener la exclusividad en diez años. La razón es que su exposición a “Precios de Referencia Externos” y comercio paralelo se intensificaría al aumentar los países de lanzamiento, con la correspondiente presión a la baja en los precios. Si no esperaran beneficios más bajos en la nueva situación (igual incentivo, pero más países de lanzamiento que antes) ya se habrían comercializado en más EE. MM. antes del cambio legislativo. Los costes administrativos emergentes superarían a los beneficios de comercializar en más países (Kyle et al., 2023).
La nueva regulación tiene que proseguir su tramitación, ahora con nueva Comisión y Parlamento, y puede haber algunos cambios. En todo caso, el objetivo a alcanzar es un equilibrio entre promoción de la innovación y posibilidades de acceso asequible a las nuevas terapias por parte de los sistemas de salud.
Notas
* Universidad Carlos III y Funcas.
1 Los EE. UU. introdujeron antes regulaciones de este tipo, aunque difieren de las europeas en algunos aspectos.
2 Con dos años adicionales en favor de los medicamentos y las indicaciones pediátricas.
3 Comienzan cuando se registran, en una fase muy temprana de desarrollo del producto, pero no son efectivas hasta que se obtiene la autorización sanitaria de comercialización, cuya fecha es a priori incierta (Kyle, 2023).
Referencias
Becker, R. (2024). The EU pharmaceutical reform. Jornada de estudio presencial. Regulación económica de la industria farmacéutica. Situación actual y perspectivas de futuro en España. 25 de abril. Funcas. https://www.funcas.es/wp-content/uploads/2024/04/Rainer-Becker.pdf
Castanheira, M., y Siotis, G. (2023). Stimulating orphan disease research: from low-cost repurposing to pricey nichebusting. Working Paper, 23-6. Difusion. Université Libre de Bruxelles. https://difusion.ulb.ac.be/vufind/Record/ULB-DIPOT:oai:dipot.ulb.ac.be:2013/368286/Holdings
CITELINE. (2024). Pharma R&D. Annual Review 2024. Supplement: New active substances launched during 2023. https://www.citeline.com/pharma-rd?utm_source=google&utm_medium=cpc&utm_term=pharma%20industry%20review%7C&utm_campaign=-ClinicalPharmaRD24EMEA&gad_source=1&gbraid=0AAAAApYLuD6JD_KiYVsFzkjDKHpjONaTl&gclid=Cj0KCQjwj4K5BhDYARIsAD1Ly2r1gkGNicG91Vj8QxHW1ggtlBsYZtS9ctjGY7FU642EP-S-p8OxSIaAt8oEALw_wcB
Comisión Europea. (2020a). Comunicación de la Comisión al Parlamento Europeo, al Consejo, al Comité Económico y Social Europeo y al Comité de las Regiones. Estrategia Farmacéutica para Europa. Bruselas, 25.11.2020. COM(2020) 761 final. SWD(2020) 286 final. https://health.ec.europa.eu/medicinal-products/pharmaceutical-strategy-europe_es
Comisión Europea. (2020b). Commission staff working document. Evaluation. Joint evaluation of Regulation (EC) No 1901/2006 of the European Parliament and of the Council of 12 December 2006 on medicinal products for paediatric use and Regulation (EC) No 141/2000 of the European Parliament and of the Council of 16 December 1999 on orphan medicinal products. Brussels, 11.8.2020. SWD(2020) 163 final. PART 1/6.
Comisión Europea. (2023a). Comunicación de la Comisión al Parlamento Europeo, al Consejo, al Comité Económico y Social Europeo y al Comité de las Regiones. Reforma de la legislación farmacéutica y medidas contra la resistencia a los antimicrobianos. COM (2023) 190 final, 26 April 2023. https://eur-lex.europa.eu/legal-content/EN/TXT/?uri=CELEX%3A52023DC0190&qid=1682665765572
Comisión Europea. (2023b). Propuesta de Directiva del Parlamento Europeo y del Consejo por la que se establece un código de la Unión sobre medicamentos para uso humano y por la que se derogan la Directiva 2001/83/CE y la Directiva 2009/35/CE. Bruselas, 26.4.2023. COM(2023) 192 final. 2023/0132.
Comisión Europea. (2023c). Propuesta de Reglamento del Parlamento Europeo y del Consejo por el que se establecen los procedimientos de la Unión para la autorización y el control de los medicamentos de uso humano, se establecen las normas por las que se rige la Agencia Europea de Medicamentos, se modifican el Reglamento (CE) n.º 1394/2007 y el Reglamento (UE) n.º 536/2014 y se derogan el Reglamento (CE) n.º 726/2004, el Reglamento (CE) n.º 141/2000 y el Reglamento (CE) n.º 1901/2006. Bruselas, 26.4.2023. COM(2023) 193 final. 2023/0131 (COD Comisión Europea).
Comisión Europea. (2023d). Commission staff working document. Impact assessment report accompanying the documents: Proposal for a Directive… on the Union code relating to medicinal products for human use…; Proposal for a Regulation… laying down Union procedures for the authorisation and supervision of medicinal products for human use and establishing rules governing the European Medicines Agency… Brussels, 26.4.2023 SWD(2023) 192 final. https://health.ec.europa.eu/document/download/027a1084-0 y https://health.ec.europa.eu/document/download/027a1084-0540-4bb6-b669-aa6cf3887684_en?filename=swd_2023_192_1_ia_ en.pdf
Comisión Europea. (2024). EU Pharmaceutical Reform: Incentives to steer innovation and achieve public health objectives. Factsheet. February.
Deaton, A. (2015). El gran escape. Salud, riqueza y los orígenes de la desigualdad. Fondo de Cultura Económica. Mexico. V. española de la inglesa de 2013. Princeton University Press.
de la Torre, B. G., y Albericio, F. (2024). The pharmaceutical industry in 2023: An analysis of FDA drug approvals from the perspective of Molecules. Molecules 2024, 29, 585. https://doi.org/10.3390/ molecules29030585
Dirección General de Cartera de Servicios del SNS y Farmacia (DGCSSNSF). Ministerio de Sanidad de España. Informe de evolución de la financiación y fijación de precio de los medicamentos huérfanos en el SNS (2016-2022).
European Federation of Pharmaceutical Industry Associations (EFPIA). (2024). The Pharmaceutical Industry in figures. Key Data 2024.https://efpia.eu/media/2rxdkn43/the-pharmaceutical-industry-in-figures-2024.pdf
European Medicines Agency (EMA). (2023). Orphan drugs in the EU. Factsheet. https://www.ema.europa.eu/en/documents/leaflet/leaflet-orphan-medicines-eu_en.pdf
Gaessler, F., y Wagner, S. (2022). Patents, Data exclusivity, and the development of new drugs. The Review of Economics and Statistics, 104(3), 571–586. doi: https://doi.org/10.1162/rest_a_00987
Horrow, C., y Kesselheim, A. S. (2023). Confronting high costs and clinical uncertainty: innovative payment models for gene therapies. Health Affairs, Noviembre, 42-11. https://www.healthaffairs.org/doi/10.1377/hlthaff.2023.00527
Kyle, M. K. (2022). Incentives for pharmaceutical innovation: What’s working, what’s lacking. International Journal of Industrial Organization, 84, 102850. https://www.sciencedirect.com/science/article/pii/S0167718722000261?via%3Dihub
Kyle, M., Corus, S., y Tanndal, J. (2023). The economics of new product launches and access to pharmaceutical products in the EU: A perspective on the EC’s proposed reform of the EU pharmaceutical legislation. Concurrences, Nº 3.
Lobo, F. (2019). La economía de la I+D en la industria farmacéutica: un resumen. Papeles de Economía Española, 190. https://www.funcas.es/wp-content/uploads/Migracion/Articulos/FUNCAS_PEE/160art05.pdf
Maini, L., y Pammolli, F. (2023). Reference pricing as a deterrent to entry: evidence from the European pharmaceutical market. American Economic Journal: Microeconomics, 15(2), 345–383.
Moa, A. (2023). 2023 New Drug Approvals: Review of New FDA and EMA Marketing Authorisations. Tribeca Knowledge. https://www.tribecaknowledge.com/blog/2023-new-drug-approvals-review-of-new-fda-and-ema-marketing-authorisations
Papapetropoulos, A., Topouzis, S., Alexander, S. P. H., et al. (2024). Novel drugs approved by the EMA, the FDA, and the MHRA in 2023: A year in review. British Journal of Pharmacology, 181(11). https://doi. org/10.1111/bph.16337
Sarpatwari, A., Beall, R. F., Abdurrob, A., He, M., y Kesselheim, A. S. (2018). Evaluating the impact of the orphan drug act’s seven-year market exclusivity period. Health Affairs, 37(5), 732–737. doi: 10.1377/ hlthaff.2017.1179. PMID: 29733729
Sumario
- Carta de la redacción
- Perspectivas económicas y fiscales para España 2024-2025
- La reforma de la financiación autonómica y el pacto catalán
- Presupuesto de 2025: en punto muerto por el acuerdo de financiación en Cataluña
- La producción industrial en la zona del euro: debilidades y desafíos
- Nuevo manual monetario
- Capital contracíclico en la banca española: su revisión en el marco de los colchones de capital
- Las exportaciones españolas de productos de alta tecnología: evolución hasta 2023
- Brecha salarial de género en España
- Los incentivos a la I+D+i en la reforma de la legislación europea del medicamento